
Interstitial lung disease (ILD) is recognized as one of the most frequent and potentially life-threatening pulmonary complications of systemic autoimmune rheumatic diseases (SARDs), a group of rare chronic inflammatory conditions characterized by autoimmune system dysregulation and chronic systemic inflammation. ILDs are a heterogeneous group of lung disorders that are underpinned by varying degrees of inflammation and fibrosis of the lung parenchyma.1 Known causes of ILD include environmental and occupational exposures-related ILD, particularly hypersensitivity pneumonitis, and SARD-related ILDs, such as rheumatoid arthritis (RA), systemic sclerosis (SSc), idiopathic inflammatory myopathies (IIM), systemic lupus erythematosus (SLE) and Sjögren’s syndrome (SjS). The most common ILD is idiopathic pulmonary fibrosis (IPF), which is an unknown cause of ILD, but incidence and prevalence vary in different locations, with some describing SARD-ILD as the most frequent form of ILD.1,2 The disease course of ILD is extremely variable, ranging from mild to rapidly progressive and potentially life-threatening respiratory failure. Its presence is a significantly negative prognostic indicator, associated with increased mortality and morbidity rates among patients with SARDs.3 It not only greatly affects patient quality of life but also poses a significant economic burden, by incurring large healthcare–related and societal–based costs.
Epidemiological data show high variability in incidence and prevalence rates of SARD-ILD, as rates vary significantly depending on the underlying rheumatic disease.4 Although ILD manifests via a vast range of SARDs, there are some conditions that display particularly higher incidences, therefore necessitating careful routine baseline and follow-up assessments, such as RA, SSc and some subgroups of IIM, such as antisynthetase syndrome and overlap syndromes (e.g. polymyositis/scleroderma [PM/Scl], anti-Ku protein antibody or anti-U1 ribonucleoprotein antibodies [U1RNP]).5 ILD is less commonly associated with SLE, anti-neutrophil cytoplasmic antobody (ANCA) associated vasculitis (AAV; particularly microscopic polyangiitis), mixed connective tissue disease (MCTD) and primary SjS. The prevalence rate of SARD–related ILD is said to be approximately 40%, with the highest rates in SSc and the lowest in patients with SLE.6 A 2023 meta-analysis of pooled prevalence revealed that ILD is estimated to affect 39–72% of patients with MCTD, 44–50% with SSc, 33–50% with IIM, 12–21% with primary SjS and 3–10% with SLE.4 Rates have increased in recent years due to improved diagnostics; however, it is still thought to be widely underdiagnosed and undertreated.5 Registry data might help in providing insights into prevalence rates; however, worldwide discrepancies are still evident.4,6,7
SARD-ILD pathogenesis is multi-faceted and poorly understood. It is believed to be due to an interplay of genetic and environmental triggers, coupled with chronic inflammation, immune dysregulation and a loss of self-tolerance. Excessive alveolar injury due to aberrant immune responses and an influx of immune cells, driven by cellular mediators such as interleukin (IL)-6, transforming growth factor beta and type 1 interferons, triggers an inflammatory cascade.1 This results in the over-activation and proliferation of fibroblasts, the cells responsible for creating connective tissue and triggers their differentiation into myofibroblasts.8 These myofibroblasts drive excessive extracellular matrix protein deposition, which causes lung scarring, fibrosis and eventually tissue remodelling due to irreversible damage to the alveolar interstitium.9
There are considerable discrepancies between what some researchers consider risk factors for developing SARD-ILD; however, some generally include advanced age, male gender, usual interstitial pneumonia (UIP) pattern and extensive lung involvement at diagnosis.10 Different SARD subtypes are associated with different risk profiles. For example, male gender, smoking and the presence of anti-cyclic citrullinated peptide antibodies are considered risk factors for RA-ILD, whereas in SSc-ILD, risk factors include diffuse cutaneous SSc, positive anti-topoisomerase 1 antibodies (Scl-70) and male gender again.4,11,12
Despite advancements, challenges remain in detecting and predicting disease progression. Most high-quality evidence on this topic comes from randomized controlled trials (RCTs) on SSc, for example, the Scleroderma Lung Study I and II (Mycophenolate vs. Oral Cyclophosphamide in Scleroderma Interstitial Lung Disease [The Scleroderma Lung Study II] [ SLSII]; ClinicalTrials.gov identifier: NCT00883129), with a notable scarcity of good-quality research available for other SARDs.13,14 Although SARDs exhibit some shared mechanisms, their heterogeneity highlights the need for improving research across all SARD-ILD types, as well as refining screening, diagnostic tools and monitoring protocols for disease-specific pathways. The development of reliable biomarkers and the use of novel personalized therapeutic approaches are also fundamental. A deeper understanding of the disease-specific mechanisms driving ILD in these frequently affected SARDs will ultimately enhance therapeutic strategies, as systemic disease activity heavily influences treatment approaches. For example, IIM-ILD can be more rapidly progressive when compared with SSc-ILD, and treatment strategies should reflect this. Additionally, two recent guidelines from the main respiratory and rheumatological societies, the American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) guidelines and the European Respiratory Society (ERS)/European Alliance of Associations for Rheumatology (EULAR) guidelines, show discrepancies between them, which also highlights a lack of certainty on the topic.15,16 This shows the need for further comparative trials to clarify the most effective management pathways, even for established treatments, such as corticosteroids. Early detection, intervention and shared decision-making between pulmonologists and rheumatologists are critical for optimizing patient outcomes and preventing unidentified inflammation that drives irreversible fibrosis, respiratory failure and premature death.
This article aims to provide an overview of the current therapeutic landscape of SARD-ILD and to discuss the current unmet needs in relation to its screening, diagnosis and monitoring.
Clinical presentation and diagnosis
The diagnosis and management of SARD-ILD are complex due to its heterogeneity and often insidious onset. Its clinical presentation is dependent on the underlying rheumatic disease and ILD subtype. ILD can appear during initial presentation, during the course of established disease or as idiopathic pneumonia with autoimmune features (IPAFs) in patients without a clearly defined SARD.17,18 ILD should always be considered in patients with SARD and requires a high index of suspicion 15, especially when respiratory symptoms are evident. Symptoms can often be absent in subclinical ILD and may only be identified via screening investigations. In some SARDs, such as RA, the concern is that ILD can remain clinically silent until advanced stages of disease, leading to significant diagnostic delays, which correlate with poorer patient outcomes 17. In patients with established SARD, ILD may develop as a secondary complication resulting from prolonged systemic inflammation and persistent immune-mediated tissue injury. Early identification of ILD in these patient populations can effectively guide their therapeutic management, making regular respiratory monitoring effective for identifying early changes, such as the presence of ‘velcro-type’ crackles, which most frequently correlate with pulmonary fibrosis. 19
Pulmonary ILD involvement can also precede systemic symptoms in up to 25% of cases.16 Guidelines advocate screening for symptoms and examination that may point to a SARD diagnosis, such as dry eyes, dry mouth, joint stiffness/swelling, Raynaud’s phenomena or mechanic’s hands, as well as performing serological autoantibody testing in all patients presenting with ILD.16
Some cases of ILD might not fulfil established criteria for a specific SARD, but still retain characteristics that point out to autoimmunity as the probable aetiology. These patients with a ‘flavour’ of autoimmunity usually have specific morphological patterns of disease, such as non-specific interstitial pneumonia (NSIP) or organizing pneumonia; clinical features of autoimmune diseases, such as mechanic’s hands and/or positive autoantibodies, such as high titres of antinuclear antibodies (ANA) or specific autoantibodies. They have been collectively termed ‘IPAF’, but, due to lack of evidence and consensus, this is still only considered a research term and, therefore, will not be addressed here in detail.18
Diagnosis is currently established via an individualized multidisciplinary approach, which is considered the gold standard for all ILD diagnosis. Investigations for suspected ILD typically include a chest X-ray, although this usually has low sensitivity in early disease, and pulmonary function testing (PFT) with spirometry and diffusing lung capacity for carbon monoxide (DLCO) (Table 1).15,16 Fibrotic ILD tends to result in a restrictive ventilatory defect, with reduced forced vital capacity (FVC), reduced total lung capacity (TLC) and reduced DLCO. DLCO may fall more significantly despite preserved FVC and TLC in early disease. A diagnosis of pulmonary arterial hypertension should be explored when there is a significantly disproportionate decline in DLCO in comparison to TLC/FVC, particularly in the context of SSc.20 High-resolution computed tomography (HRCT) of the chest provides the greatest sensitivity for detecting small interstitial abnormalities, making it essential for establishing the diagnosis of ILD. HRCT additionally allows for the quantification of parenchymal involvement, dominant process identification (fibrotic versus ground-glass), radiological pattern recognition (UIP versus non-UIP) and evaluation of disease progression.21,22
Table 1: Recommended screening and monitoring approaches according to guidelines from ACR/CHEST and ERS/EULAR15,16
| Recommendation | ACR/CHEST15 SARD-ILD | ERS/EULAR16 CTD-ILD |
| Screening tools | HRCT and PFTs recommended. HRCT alone is recommended for diagnosis | HRCT preferred. PFTs and lung ultrasound should not replace HRCT for screening |
| Screening approach | Recommends PFTs and HRCT for those at increased risk within SARD groups; not all RA and SjD require screening | Recommends HRCT for all SSc and MCTD, HRCT for IIM with risk factors (conditional for all); HRCT for RA and SjD with risk factors. No recommendation for SLE |
| Monitoring tools | PFTs (spirometry, lung volumes, DLCO) and HRCT. Also include the ambulatory desaturation test in the recommendation. Do not recommend chest X-ray, 6MWT and bronchoscopy for monitoring | PFTs (FVC, DLCO) and HRCT. PROMs and 6MWT used in addition |
| Monitoring frequency | PFTs every 3–6 months for SSc and IIM. PFTs every 3–12 months for RA, SjS and MCTD initially, then less frequent once stable. Conduct HRCT when clinically indicated | PFTs done every 3–6 months initially, then every 6–12 months. HRCT every 1–2 years for SSc, RA, SjS, SLE and MCTD. IIM may require more frequent HRCT if considered high risk |
ERS/EULAR uses the term CTD-ILD, which has been referred as SARD-ILD during this review.
ACR/CHEST = American College of Rheumatology/American College of Chest Physicians; CTD-ILD = connective tissue disease-associated interstitial lung disease; DLCO = diffusion lung capacity of carbon monoxide; ERS/EULAR = European Respiratory Society/European Alliance of Associations for Rheumatology; FVC = forced vital capacity; HRCT = high-resolution computed tomography; IIM = idiopathic inflammatory myopathies; MCTD = mixed connective tissue disease;6MWT = six-minute walking test; PFT = pulmonary function tests; PROMs = patient-reported outcome measures; RA = rheumatoid arthritis; SARD-ILD = systemic autoimmune rheumatic diseases-associated interstitial lung disease; SjD = Sjogren’s disease; SjS = Sjögren’s syndrome; SLE = systemic lupus erythematosus; SSc = systemic sclerosis.
HRCT patterns are variable in SARD-ILD, and some patterns are more prominent in certain SARD subgroups. For example, a UIP pattern, characterized by subpleural bibasal reticulation, traction bronchiectasis and honeycombing with minimal ground glass, is more frequent in RA.4 Conversely, an NSIP pattern, characterized by subpleural bibasal reticulation and ground-glass opacities, is more frequent in SSc and IIM (Figure 1).4 However, ILD patterns are not exclusive to a particular SARD and can occur in all SARD-ILDs at varying frequencies. Given the strong correlation between SARD and ILD radiological and histopathological findings, histological confirmation via biopsy is seldom required, unless alternative diagnoses, such as malignancy, hypersensitivity pneumonitis or pneumoconiosis, are being considered. Depending on the clinical context, bronchoalveolar lavage may be used to narrow the differential diagnosis, such as when infection is suspected, or there is a history of relevant exposures.16
Figure 1: Usual interstitial pneumonia and a nonspecific interstitial pneumonia pattern
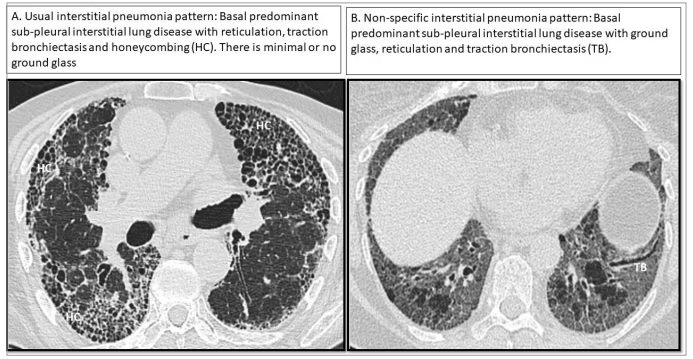
Representative high–resolution computed tomography imaging showing a usual interstitial pneumonia (A) and a nonspecific interstitial pneumonia pattern (B). Images are from a personal archive
Screening and monitoring strategies
Screening for SARD-ILDs is a key part of patient care, yet up until recently, there were no official guidelines in the UK or the USA for screening high-risk patients. It was not until 2023 that the first full-length guideline for screening and monitoring patients with SARD-ILD was published by the ACR and CHEST (Figure 2).15 It included recommended screening and monitoring strategies for people with SARDs, including RA, SSc, IIM, MCTD and SjS, but with extra emphasis for those with SSc and IIM due to their higher risk of ILD development compared with the other subtypes.5,15 As for specific screening protocols, ACR recommends PFT, which includes spirometry, lung volumes and diffusion capacity alongside HRCT.15 HRCT is much more sensitive and specific in detecting ILD compared with radiography or other imaging modalities.23 Due to the high incidence of ILD in SSc and IIM, the ACR guideline recommends screening all patients with SSc and IIM at diagnosis; however, for other SARDs, they recommend a risk factor-driven approach.15 SSc specifically requires HRCT at the time of SSc diagnosis and then annually, with PFTs including spirometry, lung volumes and DLCO. As for determining which patients require screening or not, much of the judgement is placed upon the ordering clinician.15
Figure 2: Screening and monitoring recommendations for SARD-ILD15
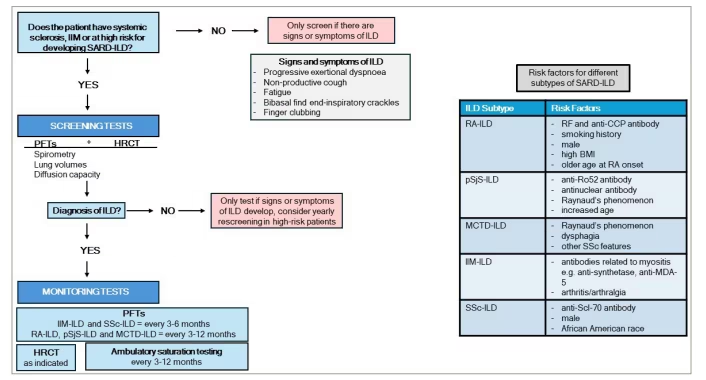
Schematic algorithm depicting the screening and monitoring recommendations for interstitial lung disease in systemic autoimmune rheumatic diseases
HRCT = high-resolution computed tomography; IIM = idiopathic inflammatory myopathies; ILD = interstitial lung disease; MCTD = mixed connective tissue disease; PFT = pulmonary function testing; pSjS = primary Sjögren’s syndrome; RA = rheumatoid arthritis; SARD = systemic autoimmune rheumatic disease; SSc = systemic sclerosis.
A newly released guideline from the ERS and EULAR provides comprehensive, evidence-based recommendations for the screening and management of connective tissue disease-associated ILDs, including SSc, RA, IIM, SjS, SLE and MCTD.16 Published in September 2025, this guideline seeks to standardize key aspects for both the treatment and the screening/monitoring of SARD-ILD. The ERS/EULAR recommendations on screening and monitoring closely align with the previously discussed ACR/CHEST guidelines, but a detailed comparison is provided in Table 1.
As for RA specifically, the most prevalent SARD in the adult population, there is currently no general consensus on which patients should undergo ILD screening. As a result, a clinical presentation and risk factor–based approach is most commonly used. There are several screening tools in development, including the Étude et Suivi des Polyarthrites Indifférenciées Récentes (ESPOIR) model, the Autoantibodies, Non-articular manifestations of RA, Cigarettes, He/him, Older age at RA onset, RA high disease activity (ANCHOR-RA) criteria, the Sociedad Española de Reumatología (SER) and Sociedad Española de Neumología y Cirugía Torácica (SEPAR) SER-SEPAR criteria, the Paulin criteria and the Four-Factor score, among others, that can be applied to detect subclinical RA-ILD.24–28 Each of the tools has differing demographic, disease activity-related and serological criteria, with varied weighting contributing to the model for RA-ILD detection.24–29 The Juge et al. ESPOIR risk scoring system was developed to assist clinicians in identifying patients who are more likely to develop RA-ILD, especially when asymptomatic.24 The cross-sectional research considered various factors, but ultimately focused on four key variables: male sex, older age at RA onset, RA disease activity measured with disease activity score-28 using erythrocyte sedimentation rate (ESR) and the Mucin-5B (MUC-5B) genetic variant associated with RA-ILD.24 The scoring system’s performance notably improved with the inclusion of the genetic variant, potentially indicating a future focus on genetic studies in ILD research. The ANCHOR-RA multi-centre study aims to enrol 1,200 patients with RA to develop a risk model of developing RA-ILD.25 The model will incorporate routinely collected clinical data, including patient demographics, RA–related characteristics (e.g. family history and inflammatory markers), comorbidities, medication, respiratory assessments (lung function and HRCT) and patient–reported outcomes, to develop a probability score for RA-ILD, which can inform further guidelines such as ACR/CHEST and ERS/EULAR.25 The SER-SEPAR model includes weighted elements of gender, age, smoking, disease duration, activity, family history and autoimmune status.26 The model has been externally validated in 146 patients with early ILD, with a sensitivity of 92.9%, specificity of 45.8% and diagnostic accuracy of 54.8%.26 The Paulin score incorporates gender, smoking status, ESR and disease activity in a weighted score.27 In a prospective Study of Inflammatory Arthritis and Interstitial Lung Disease in Early RA (SAIL-RA), the predictive performance of six screening tools for detecting RA-ILD was compared in a cohort of 172 patients with RA.30 The SER-SEPAR, ANCHOR-RA and ESPOIR showed the greatest performance, with area under the curve (AUC) of 0.82, 0.81 and 0.8, respectively. Conversely, the Four-Factor score and Paulin score performed less well, with AUCs of 0.77 and 0.6, respectively.30 Risk-based screening is a common recommendation put forward by clinicians/researchers and in current ACR guidelines.
Once a patient is diagnosed with SARD-ILD based on clinical history, signs, symptoms, PFTs and HRCT findings, the ideal monitoring plan can be put into place. PFTs are a key element of the monitoring strategy set by ACR guidelines, and the frequency varies according to the subtype of SARD. PFTs are suggested to be performed every 3–6 months for those with myositis-related ILD and SSc-ILD, whereas those with RA, MCTD and primary SjS-ILD are recommended to undergo PFTs every 3–12 months. HRCT is offered as clinically indicated, and ambulatory desaturation tests are recommended every 3–12 months (Figure 2).15 It is important that baseline PFTs and HRCT are established to ensure that ILD progression can be accurately assessed, but it is also key to include a multidisciplinary team (MDT) to ensure adequate follow-up for each patient. This MDT should include rheumatologists, respiratory clinicians, radiologists and pathologists, among others.
Looking to the future of SARD-ILD guidelines, the British Society of Rheumatology (BSR) is currently creating the UK’s first full-length guideline dedicated to SARD-ILD diagnosis and management.31 Current practice in the UK varies, and it is believed this guideline will allow healthcare providers to synthesize the current information spread across other medical guidelines and current validated research. The upcoming BSR guidelines will cover people with ILD associated with RA, SSc, SjS, IIM, MCTD, SLE, AAV and ankylosing spondylopathies (AxSpA). The BSR guideline scope also includes possible complications that should be monitored, such as pulmonary hypertension (due to ILD), gastro-oesophageal reflux disease and progressive fibrosis.31
Limitations/challenges of screening and monitoring SARD-ILD
An obvious limitation for the screening and monitoring of SARD-ILD is the limited evidence base for who should be screened and the optimal frequency of screening. The purpose of a screening programme is to identify ILD at the earliest opportunity with the aim of instituting strategies to prevent ILD progression. ILD can be a frequent complication in RA, as high as 18.7%.32 However, only 32% of patients with RA-ILD will have significant progressive disease at 5 years.33 Our ability to predict ILD progression is limited, and current research is focused on developing blood or imaging biomarkers that may be more sensitive in predicting progression than our current broad strategy of monitoring all patients once ILD is detected. A broad-brush approach of screening all patients with SARD for ILD or monitoring all SARD-ILDs with PFTs and HRCT at regular intervals once ILD is detected is resource-intensive and not economically viable. We need to develop more sophisticated, tailored tools to risk stratify patients so that resources are tailored to individuals most at risk of progression and thus investigations are appropriately utilized.
The impact of early treatment of ILD once detected is also not evident. Treatments can have adverse effects, and one needs to balance these potential negative risks with any positive impact of treatment. No study to date has evaluated whether early treatment of SARD-ILD is of benefit in preventing progression or in impacting subsequent morbidity and mortality. Furthermore, there are a number of potential treatment options, from immunosuppressive to antifibrotic agents, and we lack robust evidence of their efficacy in early SARD-ILD disease. There remain a number of unanswered questions regarding the optimal screening strategies and modalities, as well as the effectiveness of therapies, if any, for early SARD-ILD. A phase III clinical trial of nerandomilast, an inhibitor of phosphodiesterase 4B, with anti-inflammatory and antifibrotic effects, in stable or progressive SARD-ILD established on immunosuppressive therapies, is currently recruiting (A Double Blind, Randomised, Placebo-controlled Trial Evaluating the Efficacy and Safety of Nerandomilast Over at Least 26 Weeks in Patients With Systemic Autoimmune Rheumatic Diseases Associated Interstitial Lung Diseases [SARD-ILD]; ClinicalTrials.gov identifier: NCT06806592).34
Current immunomodulatory treatments
Despite SARD-ILD being a heterogeneous disease, its treatment is rather homogeneous in its approach. The mainstay of therapy is the use of immunosuppressive therapies targeted against the underlying inflammatory pathogenesis. The evidence base for individual SARD-ILDs is limited, and much of the data for immunosuppressive use are from low-quality, observational cohort data or are extrapolated from the efficacy demonstrated in RCTs of SSc-ILD. Ideally, therapies should be individually tailored towards the specific ILD subtype, with targeted therapies to gain maximal efficacy with minimal adverse effects.
Both the ACR/CHEST and the ERS/EULAR provide well-defined guidelines for the first-line management of various subgroups of SARD-ILDs (Table 2).15,16 The first-line treatment recommendations for the management of SSc-ILD include mycophenolate mofetil (MMF) and tocilizumab in both guidelines. ACR/CHEST, however, strongly recommends against glucocorticoid add-on therapy in this subgroup, while ERS/EULAR also recommends avoiding it, but only in higher doses. For RA-ILD, rituximab (RTX), azathioprine (AZA) and MMF are recommended. In ERS/EULAR, specifically, however, pirfenidone is recommended for the treatment of RA-ILD in patients with a UIP pattern or with progressive pulmonary fibrosis behaviour.16 Calcineurin inhibitors and RTX are recommended for the management of IIM-ILD, with the conditional recommendation of janus kinase (JAK) inhibitors (JAKinibs) for the more rapidly progressive ILD patterns, particularly anti-melanoma differentiation-associated gene (anti-MDA5). The option of MMF, cyclophosphamide (CYC), AZA, RTX and glucocorticoids is given for the treatment of all other SARD-ILDs in both guidelines.15,16
Table 2: Brief description of first-line treatment recommendations from the ACR/CHEST and ERS/EULAR guidelines15,16
|
| SSc-ILD | RA-ILD | IIM-ILD | Other SARD-ILDs |
| ACR/CHEST15 | MMF Nintedanib Tocilizumab | RTX AZA MMF | CNI | MMF CYC AZA RTX Glucocorticoids |
| Strong recommendation against glucocorticoids | JAKi add-on | |||
| ERS/EULAR16 | MMF Nintedanib Tocilizumab | RTX AZA MMF | CNI | MMF CYC AZA RTX Glucocorticoids |
| Pirfenidone in PPF | JAKi add-on |
ACR/CHEST = American College of Rheumatology/American College of Chest Physicians; AZA = azathioprine; CNI = calcineurin inhibitors; CYC = cyclophosphamide; ERS/EULAR = European Respiratory Society/European Alliance of Associations for Rheumatology; IIM-ILD = idiopathic inflammatory myopathies-associated interstitial lung disease; JAKi = JAK inhibitors; MMF = mycophenolate mofetil; PPF = progressive pulmonary fibrosis; RA-ILD = rheumatoid arthritis-associated interstitial lung disease; RTX = rituximab; SARD-ILD = systemic autoimmune rheumatic diseases-associated interstitial lung disease; SSc-ILD = systemic sclerosis-associated interstitial lung disease.
Methotrexate
Methotrexate (MTX) is a disease-modifying antirheumatic drug used as a first-line treatment for RAdue to its effectiveness at reducing morbidity.15,16 However, there has been some apprehension regarding its use in the management of SARD-ILDs due to its putative association with lung pathology in patients with RA. A previous meta-analysis of 22 RCTs, which included 8,584 participants, found MTX treatment to be associated with increased pulmonary adverse effects than other disease-modifying anti-rheumatic drugs and biologics.35 In contrast, however, more recent studies have reported that MTX is not related to an increased risk of RA-ILD in patients with RA, and that its effect might even be protective.36 A case-control study followed 410 patients with RA-ILD and 673 control patients with RA but no ILD.36 They analysed the numbers of patients who never had MTX exposure and those who did. It was found that there was an inverse relationship between MTX exposure and RA-ILD (p=0.022). They also discovered that in patients with RA-ILD, ILD was detected significantly later in MTX–exposed patients than in those never exposed to MTX. The issue arises from the fact that MTX is generally not regarded as an effective treatment for RA-ILD, and most immunosuppressive agents used to treat ILD are considered ineffective for treating articular rheumatological manifestations.
Mycophenolate mofetil
The most robust evidence to date is in SSc-ILD. In the Scleroderma Lung Study 1 (SLS1), 145 patients with SSc-ILD were randomized to receive oral CYC or placebo.13 Patients in the CYC group reported a mean 2.53% improvement in FVC compared with their placebo-treated counterparts. Subsequently, a non-inferiority study, the Scleroderma Lung Study 2, of CYC versus MMF demonstrated similar efficacy of the two treatments.14 However, a 24-month course of MMF resulted in fewer deaths and adverse effects than its 12-month CYC followed by 12-month placebo counterpart. Although there was no significant difference between MMF and CYC when the primary endpoint of the study, improvement in FVC% from baseline, was analysed, the MMF arm resulted in fewer failed treatments, and leucopenia and thrombocytopenia occurred more often in the CYC arm of the study. The authors concluded that there was greater tolerability and a better toxicity profile with MMF than CYC, and thus MMF is now considered first-line therapy in SSc-ILD.14
Tocilizumab
Tocilizumab is a monoclonal anti-IL-6 receptor-α antibody that has been shown to reduce the decline in FVC in patients with SSc-ILD. The FaSScinate phase II trial (A Phase II/III, Multicenter, Randomized, Double-blind, Placebo-controlled Study To Assess The Efficacy And Safety Of Tocilizumab Versus Placebo In Patients With Systemic Sclerosis; ClinicalTrials.gov identifier: NCT01532869) was a double-blind, controlled study in 35 sites in the UK, USA, Germany, France and Canada.37 A total of 87 patients with progressive SSc were assigned to either a tocilizumab treatment group or a placebo group. The primary endpoint of change in modified Rodnan skin score (mRSS) at 24 weeks was not different between the two groups, although there were non-significant improvements in skin thickness in the tocilizumab-treated group at 24 and 48 weeks. This was particularly evident in early diffuse SSs. Furthermore, the secondary endpoint of per cent predicted FVC reported that significantly fewer number of patients in the tocilizumab arm had a decline in per cent predicted FVC at 24 and 48 weeks (p=0.009 at week 24; p=0.0373 at week 48). In the tocilizumab arm, 3% and 10% of patients experienced a greater than 10% FVC decline at weeks 24 and 48, respectively, compared with 19% and 23% of patients in the placebo group.37
These findings in the secondary endpoints led to the phase III multi-centre RCT of tocilizumab in patients with diffuse cutaneous SSc, the FocuSSced study (A Phase III, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study to Assess the Efficacy and Safety of Tocilizumab Versus Placebo in Patients With Systemic Sclerosis; ClinicalTrials.gov identifier: NCT02453256; Figure 3).38 This study recruited patients with diffuse cutaneous SSc, moderate skin involvement with mRSS 10–35, elevated acute-phase inflammatory markers and active disease. A total of 212 participants were recruited: 107 in the placebo arm and 105 treated with tocilizumab. Participants had mild lung function impairment at baseline (FVC, >80%; DLCO, >74%) and 65% had ILD on HRCT. As in the phase II study, the primary endpoint of change in mRSS at 48 weeks was not met. At week 48, the change from baseline of per cent predicted FVC was -4.6 in the placebo arm and -0.4 in the tocilizumab group (nominal p=0.0002; Figure 3). As the primary endpoint did not reach 5% significance, all secondary endpoints were exploratory in nature. However, the lung function differences in the tocilizumab–treated group in a specific cohort of patients with early ILD and high inflammatory markers have led to the US Food and Drug Administration (FDA) approval of tocilizumab for the treatment of SSc-ILD.39
Figure 3: Key features of the FocuSSced trial38
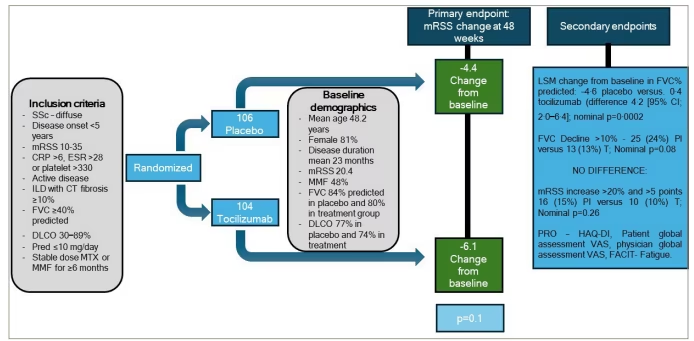
Schematic showing key features of the FocuSSced randomized controlled trial of tocilizumab in systemic sclerosis
CI = confidence interval; CT = computerised tomography; CRP = C-reactive protein; DLCO = transfer factor; ESR = erythrocyte sedimentation rate; FACIT = functional assessment of chronic illness therapy; FVC = forced vital capacity; HAQ-DI = Health Assessment Questionnaire-Disability Index; ILD = interstitial lung disease; LSM = least square mean; MMF = mycophenolate; mRSS = modified Rodnan skin score; MTX = methotrexate; Pl = placebo; Pred = prednisolone; PRO = patient-reported outcome; SSc = systemic sclerosis; T = tocilizumab; VAS = visual analogue score.
Rituximab
The rituximab versus cyclophosphomide in patients with connective tissue disease assoxiated ILD (RECITAL) study (A Randomized, Double Blind Controlled Trial Comparing Rituximab Against Intravenous Cyclophosphamide in Connective Tissue Disease Associated Interstitial Lung Disease; ClinicalTrials.gov identifier: NCT01862926), a non-inferiority RCT of RTX versus CYC in patients with progressive SARD-ILD despite conventional immunosuppressive therapy, showed RTX to be the more tolerable of the two, with less corticosteroid exposure also.40 A total of 101 participants were randomly allocated to a CYC treatment arm or an RTX arm, and findings such as FVC changes, quality-of-life, corticosteroid exposure and adverse effects were analysed. Although there was no significant difference in the primary outcome, FVC improvements, between the two arms after 24 weeks, there were numerically fewer adverse effects reported by week 48 in the RTX arm, 445, than in the CYC arm, 646. These included gastrointestinal disorders, administration-site reactions and nervous system disorders. In addition to this finding, it was also reported that the mean steroid exposure per patient over the 48-week period was 11,469 mg in the RTX group and 13,291 mg in the CYC group. Overall, this study favoured RTX over CYC in patients with SARD-ILD who were progressing despite second–line immunosuppressive therapy, posing a role for RTX as a rescue therapy in progressive SARD-ILD. SSc-ILD and IIM-ILD were the predominant SARDs in the RECITAL study.
Furthermore, the rituximab and mycophenolate mofetil combination in patients with ILD (EVER-ILD) trial (NCT02990286) prospectively evaluated the outcome of change in FVC in 122 patients with ILD with an NSIP pattern.41 The patients were randomly allocated to a treatment group receiving MMF and placebo or another group receiving MMF and RTX. Upon measuring the primary efficacy endpoint at 6 months, the MMF- and RTX-treated group recorded an increase in 1.60% of their predicted FVC, while the MMF- and placebo-treated group recorded a decrease, -2.01%, of their predicted FVC (p=0.0273). Although these results suggest that RTX and MMF combination therapy has greater efficacy than MMF alone in patients with ILD with an NSIP pattern, further clinical trials are necessary to confirm these findings and to evaluate cost-effectiveness.
Similarly, the safety and efficacy of rituximab in systemic sclerosis (DESIRSES) trial (Double-Blind, Parallel-group Comparison, Investigators Initiated Phase II Clinical Trial of IDEC-C2B8 [Rituximab] in Patients With Systemic Sclerosis; ClinicalTrials.gov identifier: NCT04274257) was a double-blind, randomized trial in which the efficacy of RTX was measured against a placebo in SSc-ILD.42 Forty-five patients participated, of whom 25 received RTX and 23 were placebo-treated. At 24 weeks post-commencement of treatment, the change in percentage-predicted value of FVC in the RTX group, an increase of 1.61%, was found to be significantly more positive than in the placebo group, a decrease of 6.40% (p=0.0002).
The evidence for immunosuppressive therapy in RA-ILD is less robust. Unlike SSc-ILD, where the radiological pattern is predominantly an NSIP pattern, in RA-ILD, the predominant pattern is that of a UIP pattern. In IPF, the commonest ILD of unknown cause, the prednisone, azathioprine, and N-acetylcysteine: A Study That Evaluates Response in Idiopathic Pulmonary Fibrosis (PANTHER-IPF) study (Prednisone, Azathioprine, and N-acetylcysteine: A Study That Evaluates Response in IPF; ClinicalTrials.gov identifier: NCT00650091), an RCT of prednisolone, AZA and N-acetylcysteine compared with placebo, was terminated early because of increased mortality and death in the immunosuppression arm.43 Clinicians are rightly cautious about using immunosuppression in RA-UIP without robust RCT evidence of efficacy. In observational studies, fraught with bias, patients with RA-ILD on MTX had better survival than those not treated with MTX.44 However, for RA-ILD, the evidence base for immunosuppression remains limited, and there is a great need for robust evidence-based treatments in RA-ILD.
Emerging immunomodulatory treatments
Janus kinase inhibitors
JAKinibs are a pharmacological class of immunosuppressants that act by undergoing competitive adenosine triphosphate (ATP) binding in the JAK/STAT pathway, which leads to impediment of phosphorylation of cytokine receptors and blockage of gene transcription.45 This results in decreased cytokine production as well as compromised differentiation of immune cells. Some data have shown that JAKinibs may be beneficial for the stabilization or improvement of SSc-ILD in patients. A narrative, non-systematic review that looked at seven articles published between the years 2018 and 2022 found that, when treated with JAKinibs, 93% of the 31 patients analysed showed no signs of progression in their ILD.45 Pulmonary stability was measured clinically, functionally or radiologically depending on the available data from the literature. The authors concluded that, given the sparsity of available literature, data from larger cohorts and RCTs are necessary to confirm results. Nevertheless, the targeting of inflammatory and fibrotic pathways by JAKinibs indicates that they may be a promising treatment in SSc-ILD.
Antifibrotic therapies
The antifibrotic therapies pirfenidone and nintedanib are the cornerstone of treatment for the commonest ILD, IPF, and their utility in SARD-ILD has pathogenic rationale, particularly in the more fibrotic SARD-ILDs.
Nintedanib
Nintedanib is an inhibitor of intracellular tyrosine kinases, which counteracts processes that are pivotal to pulmonary fibrosis progression. The Safety and Efficacy of Nintedanib in Systemic Sclerosis (SENSCIS) RCT (A Double Blind, Randomised, Placebo-controlled Trial Evaluating Efficacy and Safety of Oral Nintedanib Treatment for at Least 52 Weeks in Patients With Systemic Sclerosis Associated Interstitial Lung Disease [SSc-ILD]; ClinicalTrials.gov identifier: NCT02597933) of nintedanib versus placebo in 576 patients with SSc-ILD met its primary endpoint of FVC decline (Figure 4).46 There was a difference in the adjusted annual rate of change in FVC of 41.0 mL, with the nintedanib group declining by -52.4 mL per year compared with a -93.3 mL per year decline in the placebo arm. A total of 48.4% of patients were concurrently treated with MMF, and 51.9% had diffuse cutaneous SSc. Secondary endpoints of change in Rodnan skin scores and quality of life were not different between the two groups. Nintedanib does, however, have an adverse effect profile that is mainly characterized by gastrointestinal events, particularly mild or moderate diarrhoea. An in-depth analysis of the adverse effects of the SENSCIS trial found that the proportions of patients with adverse effects were similar in both groups: 98.3% in the nintedanib arm and 95.8% in the placebo arm.47 Weight loss was reported by 11.8% in the nintedanib group, with a mean change of -3.22 kg from baseline, and by 4.5% in the placebo group, with a mean change of -0.25 kg from baseline. Permanent discontinuation of the treatment due to diarrhoea was reported to be 6.9% in the patients treated with nintedanib. The adverse event profile of nintedanib in patients with SSc-ILD was consistent with its established safety and tolerability profile in patients with IPF. However, these adverse effects should be managed using symptomatic therapies and dose adjustments to aid patient compliance with the therapy.
Figure 4: Key features of the SENSCIS trial46
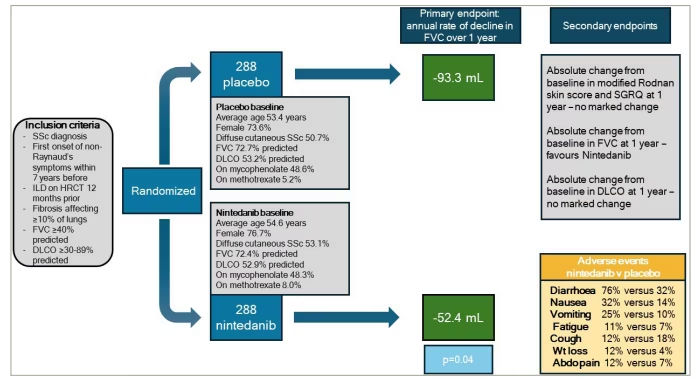
Schematic showing key features of the Safety and Efficacy of Nintedanib in Systemic Sclerosis (SENSCIS) randomized controlled trial of nintedanib in systemic sclerosis-related interstitial lung disease
Abdo = abdominal; DLCO = transfer factor; FVC = forced vital capacity; HRCT = high resolution computerised tomography; ILD = interstitial lung disease; Pred = prednisolone; SGRQ = St. George’s Respiratory Questionnaire; SSc = systemic sclerosis; Wt = weight.
Furthermore, a pre-specified subgroup analysis of SARD-ILD was explored in the nintedanib in progressive fibrosing interstitial lung disease (INBUILD) RCT (NCT02999178) of nintedanib versus placebo in patients with a diagnosis of progressive fibrosing ILD (Figure 5).48 Progressive fibrosis was defined as meeting two out of three criteria: progression in symptoms, greater than 5% FVC decline, or radiological progression over a 24-month period. A total of 663 participants were recruited to the study, of whom 170 (25.6%) had a SARD-ILD. The study found a reduction in FVC decline of 104 mL in a pre-specified SARD-ILD subgroup, with a rate of decline of -178.6 mL per year with placebo compared with -75.9 mL with nintedanib (p=0.012).48 The SARD-ILD group was a heterogeneous group comprising RA-ILD (52.4%), SSc-ILD (22.9%), MCTD-ILD (11.2%) and other SARD-ILDs (13.5%). Adverse effects were most commonly gastrointestinal, in particular diarrhoea. It was concluded that the overwhelming evidence from the trial was suggestive of the potential role of nintedanib in the treatment of SARD-ILDs for the reduction of the rate of FVC decline. Subsequently, a sub-group analysis of patients with RA-ILD from the INBUILD trial showed consistent results.49 This trial reported that, in a total of 89 patients with RA-ILD, the 52-week rate of decline in FVC was -82.6 mL in the nintedanib group versus -199.3 mL in the placebo group. The adverse outcome profile in the RA-ILD subgroup was consistent with the overall trial population, lending further evidence to the efficacy of nintedanib in patients with progressive SARD-ILD irrespective of the underlying SARD subtype. However, one needs to be mindful that the number of patients for the individual SARD subtypes is small in the INBUILD trial. Despite this, the consistent message is that, once progressive fibrosis ensues, nintedanib is efficacious irrespective of the underlying aetiology of the ILD.
Figure 5: Key features of the INBUILD trial48
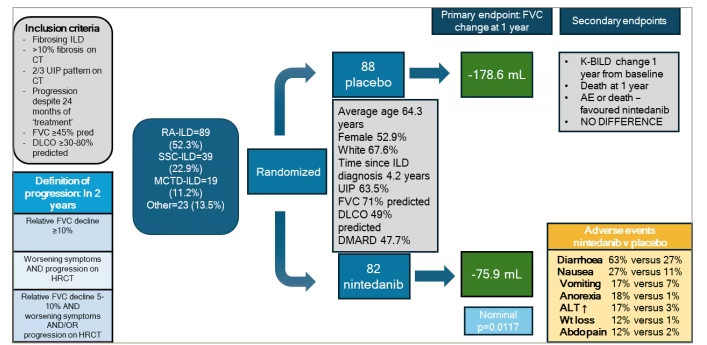
Subgroup analysis of systemic autoimmune rheumatic diseases from the nintedanib in progressive fibrosing interstitial lung disease (INBUILD) randomized controlled trial of nintedanib in progressive fibrosing interstitial lung diseases
ALT = alanine transaminase; CT = computerised tomography; Abdo = abdominal; AE = adverse event; diag = diagnosis; DMARD = disease-modifying anti-rheumatic drugs; DLCO = transfer factor; FVC = forced vital capacity; HRCT = high-resolution computed tomography; ILD = interstitial lung disease; K-BILD = King’s Brief Interstitial Lung Disease questionnaire; MCTD = mixed connective tissue disease; MCTD-ILD = mixed connective tissue disease-associated interstitial lung disease; Pred = prednisolone; RA = rheumatoid arthritis; RA-ILD = rheumatoid arthritis-associated interstitial lung disease; SSc = systemic sclerosis; SSc-ILD = systemic sclerosis-associated interstitial lung disease; UIP = usual interstitial pneumonia; Wt = weight.
Pirfenidone
Pirfenidone is an antifibrotic and anti-inflammatory agent that was approved for use in the treatment of IPF in Europe in 2011 and in the USA in 201450 . The Treatment for Rheumatoid Arthritis and Interstitial Lung Disease (TRAIL) was a phase II RCT (Phase 2 Study of Safety, Tolerability and Efficacy of Pirfenidone in Patients With Rheumatoid Arthritis Interstitial Lung Disease [TRAIL1]; ClinicalTrials.gov identifier: NCT02808871) evaluating pirfenidone therapy versus placebo therapy in RA-ILD. The trial was terminated early due to recruitment difficulties as a result of the COVID-19 pandemic.51 Despite the trial being underpowered, there remained a statistically significant reduction in the annual rate of FVC decline in the pirfenidone group than in those receiving placebo, and this effect was driven by a UIP pattern. A total of 123 patients were randomly assigned to a pirfenidone treatment group and a placebo control group. The estimated annual decline in absolute FVC was -66 mL in the pirfenidone group and -146 mL in the placebo group (p=0.008). Although limited by the reduced statistical power due to early termination, the findings do indicate that pirfenidone could potentially be beneficial for the treatment of RA-ILDs, specifically those with a UIP pattern.
Nerandomilast
Nerandomilast is a phosphodiesterase 4B inhibitor with both immunosuppressive and antifibrotic properties. The Fibroneer-IPF (A Double Blind, Randomized, Placebo-controlled Trial Evaluating the Efficacy and Safety of BI 1015550 Over at Least 52 Weeks in Patients With Idiopathic Pulmonary Fibrosis [IPF]; ClinicalTrials.gov identifier: NCT05321069) and Fibroneer-ILD (A Double Blind, Randomized, Placebo-controlled Trial Evaluating the Efficacy and Safety of BI 1015550 Over at Least 52 Weeks in Patients With Progressive Fibrosing Interstitial Lung Diseases [PF-ILDs]; ClinicalTrials.gov identifier: NCT05321082) studies have demonstrated a positive impact of nerandomilast on FVC decline in patients with IPF and progressive non-IPF fibrosis.52,53 In a subgroup analysis of patients with SARD-ILD in the FIBRONEER-ILD trial (RA, 36.3%; SSc, 23.1%; mixed, 14.5%), it was found that nerandomilast treatment reduced the annual rate of decline in FVC in patients with SARD-ILD.53 A total of 325 patients with SARD-ILD were divided into placebo, 9 mg nerandomilast twice a day (BD) and 18 mg nerandomilast BD groups. After 52 weeks, it was found that the adjusted mean change in FVC was significantly reduced with nerandomilast in this subgroup, as was seen in the overall cohort, with an FVC decline of -61.2 mL in the 9 mg nerandomilast BD group, -64.9 mL in the 18 mg nerandomilast BD group and -107.1 mL in the placebo group. Nerandomilast had a similar adverse event profile to the placebo group, and the authors concluded that this was a satisfactory safety and tolerability profile. These findings show promise for the possibility of the acceptance of nerandomilast as a mainstay of SARD-ILD treatment in the future.
Alternative treatment options: Haematopoietic stem cell and lung transplantation
Haematopoietic stem cell transplantation
It has been shown in a number of RCTs that haematopoietic stem cell transplantation (HSCT) might have greater efficacy for the treatment of SSc-ILD than CYC.54
The autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST) trial (Trial of High Dose Cyclophosphamide and Rabbit Antithymocyte Globulin [rATG] With Hematopoietic Stem Cell Support in Patients With Systemic Scleroderma: A Randomized Trial; ClinicalTrials.gov identifier: NCT00278525) measured the outcomes of 10 patients treated with HSCT versus the outcomes of nine patients treated with a 6-month regime of CYC alone.55 The patients treated with non-myeloablative, autologous HSCT also received a 200 mg/kg dose of intravenous CYC and a 6.5 mg/kg dose of intravenous rabbit antithymocyte globulin. The CYC-only-treated patients received a 1.0 g/m2 dose of intravenous CYC once every 6 months. At a 12-month follow–up, all 10 patients in the HSCT–treated group showed improved symptoms, in contrast to none in the CYC group (p=0.00001). At a follow-up 2 years after the HSCT, improvements in FVC persisted (p<0.03).
Similarly, the Autologous Stem Cell Transplantation International Scleroderma (ASTIS) trial (High dose immunoablation and autologous haematopoietic stem cell transplantation versus monthly intravenous pulse therapy; ISRCTN identifier: ISRCTN54371254) studied the efficacy of HSCT versus CYC alone in 156 patients with SSc.56 Seventy-nine patients were allocated to the HSCT group and 77 to the CYC group. They found that the mean changes from the baseline of FVC after 2 years in the HSCT group were 6.3% of predicted value, which was significantly better than the control, CYC-only group, at 2.8% (p=0.004).
Still, HSCT has been associated with high immediate morbidity and mortality, besides increasing the risk of infections, which is particularly worse in an ILD population. Therefore, centre experience usually dictates the choice to perform HSCT, and the choice of patient who might benefit the most from HSCT remains to be a research gap.
Lung transplantation
Lung transplantation (LTx) is another option for the management of SARD-ILD. In 2007, ILD surpassed chronic obstructive pulmonary disease (COPD) as the most common worldwide indication for LTx.57 The ERS strongly recommends LTx based on its reported 75% reduced risk of death in patients with IPF when compared with medical management. More research needs to be studied to statistically evaluate the efficacy of LTx in ILD; however, as the available studies are retrospective and limited by selection bias. SARD-ILD, however, is a contraindication for LTx in several centres due to extra-pulmonary involvement, such as myositis, gastro-oesophageal disease and renal disease.58 For these reasons, in the 25 years between 1995 and 2010, less than 2 in every 100 lung transplants were given to patients with SARD-ILD. Therefore, although promising, both HSCT and LTx remain viable alternative treatment options for only a minority of the SARD-ILD population.
Conclusion
To conclude, SARD-ILD is an increasingly recognized, complex clinical challenge faced by healthcare systems. It not only places a burden on the patient directly, but also on society economically. Despite recent advances, there is a considerable gap in our knowledge regarding the mechanisms driving its variability and its lack of predictability regarding onset, presentation and progression. This is evident through discrepancies in the ACR and ERS/EULAR guidelines. Enhancing research to understand what drives this underlying disease variability will ultimately prevent delays in intervention and enhance the advancement and efficacy of therapeutic options available. The therapeutic field in SARD-ILD continues to evolve, with the potential for new anti-inflammatory and antifibrotic agents to impact disease progression.
